[fullwidth background_color=”#ebebeb” background_image=”” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_repeat=”no-repeat” background_position=”left top” video_url=”” video_aspect_ratio=”16:9″ video_webm=”” video_mp4=”” video_ogv=”” video_preview_image=”” overlay_color=”” overlay_opacity=”0.5″ video_mute=”yes” video_loop=”yes” fade=”no” border_size=”0px” border_color=”” border_style=”solid” padding_top=”0″ padding_bottom=”0″ padding_left=”” padding_right=”” hundred_percent=”yes” equal_height_columns=”no” hide_on_mobile=”no” menu_anchor=”” class=”” id=””][imageframe lightbox=”no” lightbox_image=”” style_type=”none” hover_type=”none” bordercolor=”” bordersize=”0px” borderradius=”0″ stylecolor=”” align=”center” link=”” linktarget=”_self” animation_type=”0″ animation_direction=”down” animation_speed=”0.1″ hide_on_mobile=”no” class=”” id=””]  [/imageframe][/fullwidth][fullwidth background_color=”#ffffff” background_image=”” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_repeat=”no-repeat” background_position=”left top” video_url=”” video_aspect_ratio=”16:9″ video_webm=”” video_mp4=”” video_ogv=”” video_preview_image=”” overlay_color=”” overlay_opacity=”0.5″ video_mute=”yes” video_loop=”yes” fade=”no” border_size=”0px” border_color=”” border_style=”solid” padding_top=”40″ padding_bottom=”40″ padding_left=”” padding_right=”” hundred_percent=”no” equal_height_columns=”no” hide_on_mobile=”no” menu_anchor=”” class=”” id=””][two_third last=”no” spacing=”yes” center_content=”no” hide_on_mobile=”no” background_color=”” background_image=”” background_repeat=”no-repeat” background_position=”left top” border_position=”all” border_size=”0px” border_color=”” border_style=”” padding=”” margin_top=”” margin_bottom=”” animation_type=”” animation_direction=”” animation_speed=”0.1″ class=”” id=””][fusion_text]
[/imageframe][/fullwidth][fullwidth background_color=”#ffffff” background_image=”” background_parallax=”none” enable_mobile=”no” parallax_speed=”0.3″ background_repeat=”no-repeat” background_position=”left top” video_url=”” video_aspect_ratio=”16:9″ video_webm=”” video_mp4=”” video_ogv=”” video_preview_image=”” overlay_color=”” overlay_opacity=”0.5″ video_mute=”yes” video_loop=”yes” fade=”no” border_size=”0px” border_color=”” border_style=”solid” padding_top=”40″ padding_bottom=”40″ padding_left=”” padding_right=”” hundred_percent=”no” equal_height_columns=”no” hide_on_mobile=”no” menu_anchor=”” class=”” id=””][two_third last=”no” spacing=”yes” center_content=”no” hide_on_mobile=”no” background_color=”” background_image=”” background_repeat=”no-repeat” background_position=”left top” border_position=”all” border_size=”0px” border_color=”” border_style=”” padding=”” margin_top=”” margin_bottom=”” animation_type=”” animation_direction=”” animation_speed=”0.1″ class=”” id=””][fusion_text]
Pituitary Adenoma
Pituitary adenomas are tumours that occur in the pituitary gland.
Almost all are benign, but can be invasive (despite benign histological appearances), and are rarely malignant (pituitary carcinoma accounting for 0.1%-0.2% of cases). Pituitary adenomas represent between 10% and 25% of all intracranial tumours. It is estimated that the population prevalence is approximately 17%.
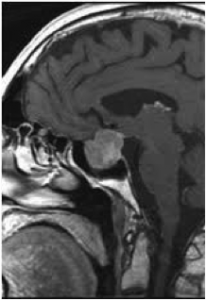 Pituitary adenomas larger than 10 mm in any dimension are classified as macroadenomas, as distinct from microadenomas, less than this size.
Pituitary adenomas larger than 10 mm in any dimension are classified as macroadenomas, as distinct from microadenomas, less than this size.
Most adenomas are microadenomas.
Many pituitary tumours are diagnosed incidentally, and, if micro and non-secretory, can be managed expectantly.
Clinically significant pituitary adenomas may present in a number of ways, the most serious of which are chiasmatic compression, causing visual field loss (bitemporal hemianopia, in which the outer parts of the visual fields are lost, contracting peripheral vision) and ultimately if untreated leading to visual failure, and pit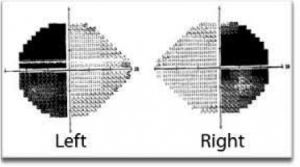 uitary hypersecretion, in which excess hormone is secreted, with unique consequences dependant on the hormone secreted.
uitary hypersecretion, in which excess hormone is secreted, with unique consequences dependant on the hormone secreted.
Pituitary adenomas are also the commonest cause of pituitary hypofunction, and pituitary failure (panhypopituitarism) is also a serious medical condition.
Hypersecretion syndromes
Prolactin hypersecretion
Occurs in males and females, but is more clinically obvious in females (galactorrhoea (lactation), amenorrhea, infertility, loss of bone density).
Potentially amenable to medical management with dopaminergic drugs (Bromocriptine , Cabergoline or Quinagolide).
Cortisone hypersecretion
The result of a pituitary tumour secreting excess, uncontrolled adrenocorticotropic hormone (ACTH), hypercortisolism results in Cushing’s disease, which, without treatment, produces significant morbidity and reduces life expectancy.
Signs and symptoms include high blood pressure, abdominal obesity, facial plethora and fullness (‘moonface’), buffalo hump, osteoporosis, ACN, muscular weakness and menstrual disturbance.
A pituitary tumour is the second most frequent cause of Cushing’s syndrome after medication-induced cases.
The most successful curative treatment is surgical, but surgical cure may be elusive, and delayed recurrences can occur.
Other strategies include medication, and radiotherapy.
Growth Hormone hypersecretion
Excess Growth Hormone (GH) results in gigantism, if occurring before skeletal maturity, or acromegaly if in adult life. Acromegaly is associated with characteristic physical changes (enlargement of hands and feet, enlargement of the tongue, facial changes including prognathism, brow enlargement, enlargement of nose, lips and ears, and general thickening of the skin. Internal organs are also affected, most notably the heart, and kidneys.
Complications due to soft tissue swelling include carpal tunnel syndrome, and weakening of the cardiac muscle, which may result in accelerated cardiovascular deterioration. Untreated acromegaly shortens life expectancy by an estimated 20%.
Optimal treatment is resection of the causative tumour. This is successful in about 60% of cases, in normalising measured growth hormone and insulin-like growth factor 1 (IGF1).
Medical options include the use of the drugs Octreotide and Lanreotide, synthetic forms of a brain hormone, somatostatin, which reduces growth hormone production.
In resistant cases, radiotherapy may be considered.
The majority of pituitary adenomas are non-secretory, and they exert their influence by their mass effect on adjacent tissues (pituitary gland, optic chiasm, hypothalamus, cavernous sinus and temporal lobe).
Pituitary hypofunction may occur, and persist despite treatment, and require replacement therapy.
Visual deterioration can result from chiasmatic compression and also from pituitary apoplexy, an acute condition involving swelling of, or bleeding into, the adenoma. Frequently this emergency situation will be heralded by sudden severe headache, with the subsequent development of visual symptoms, including field loss, double vision, and even blindness.
Frequently pituitary function will also decline, sometimes catastrophically, requiring urgent recognition and replacement.
The Neurosurgery Tasmania team have extensive experience in pituitary surgery, and work closely with ENT surgeons and endocrinologists to manage these complex tumours.[/fusion_text][/two_third][one_third last=”yes” spacing=”yes” center_content=”no” hide_on_mobile=”no” background_color=”#f7f7f7″ background_image=”” background_repeat=”no-repeat” background_position=”left top” border_position=”all” border_size=”0px” border_color=”” border_style=”solid” padding=”20px” margin_top=”” margin_bottom=”” animation_type=”0″ animation_direction=”down” animation_speed=”0.1″ class=”” id=””][fusion_widget_area name=”avada-custom-sidebar-cerebraltumours” background_color=”#f7f7f7″ padding=”10px” class=”” id=””][/fusion_widget_area][/one_third][/fullwidth]